
ALS study finds early stress signals in vulnerable neurons
A Cell study in mice and patient tissue points to early stress and protective signals in motor neurons that are prone to ALS damage.
By Tom Brennan · Health & Medicine Correspondent
3 min read
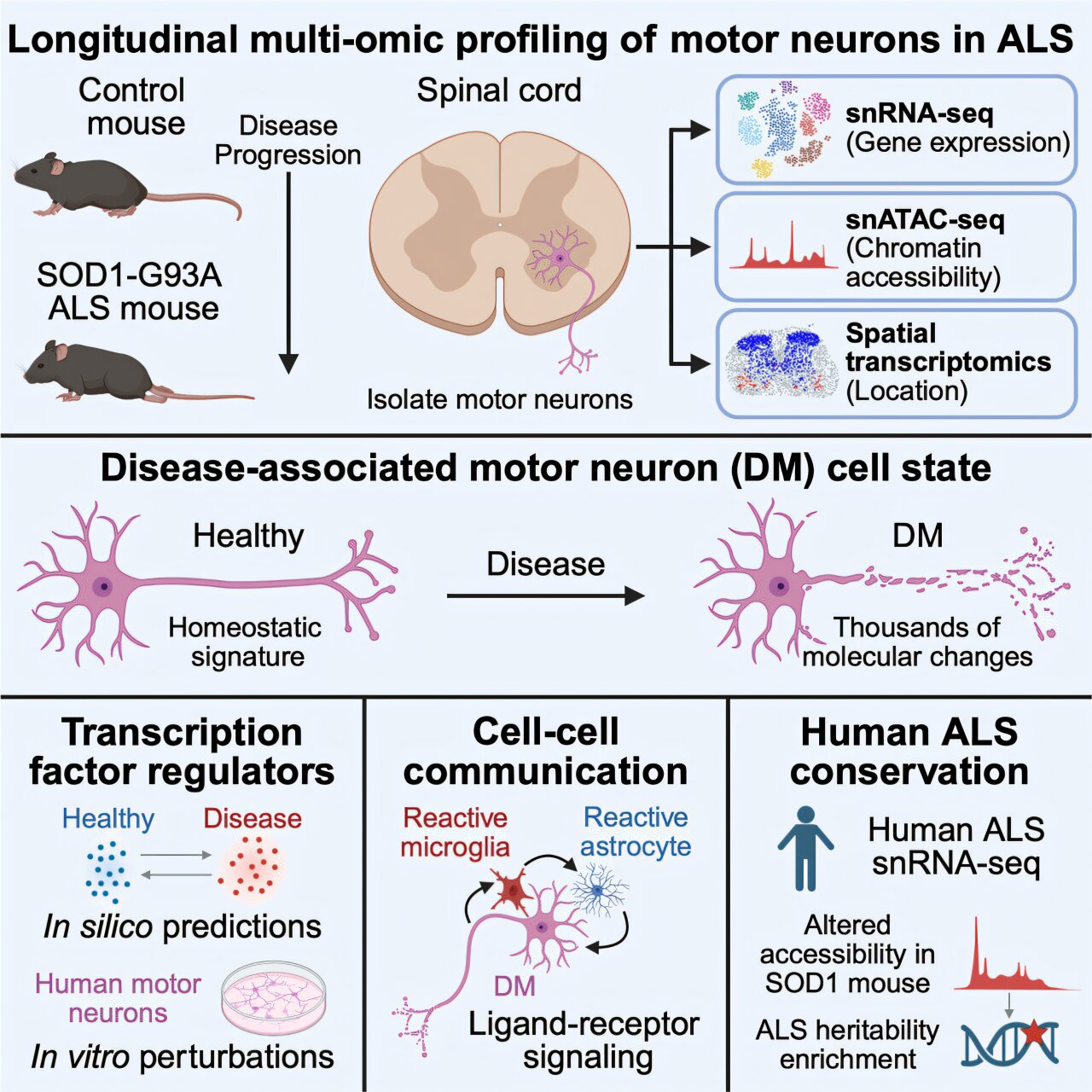
Stanford Medicine researchers have identified molecular changes that appear in vulnerable motor neurons before those cells die in amyotrophic lateral sclerosis. The findings may help explain why ALS damages some movement-controlling neurons more readily than others, according to Stanford University.
The study, published June 23, 2026, in Cell, was led by Olivia Gautier and Jacob Blum in the lab of Aaron Gitler, a Knight Initiative for Brain Resilience researcher and professor of genetics at Stanford Medicine. Gautier said the work also points to possible ways to strengthen neurons against the disease.
ALS causes motor neurons in the brain and spinal cord to break down over time, Stanford said. As the disease advances, patients lose control of movement and eventually the ability to breathe; many are diagnosed in middle to late adulthood, and most live three to five years after diagnosis, according to the university.
Tracking the neurons most at risk
Scientists have known that alpha motor neurons, which drive muscle contractions, are especially susceptible in ALS, Stanford said. The new study focused on why those neurons, and some subtypes within that group, may be more likely to fail.
The researchers used mice engineered to carry human SOD1-G93A, a disease-linked version of the SOD1 gene, according to Stanford. Mutations in SOD1 account for about 20% of inherited ALS cases, and mice carrying SOD1-G93A are widely used to study the disease.
Gautier’s team analyzed RNA in individual cells from those mice, Stanford said. That approach let the researchers compare molecular profiles across motor neuron types and look for patterns that might appear before neurons stopped functioning.
The data showed a subset of alpha motor neurons in a disease-associated state, according to the study. Those cells produced higher levels of RNA tied to stress and cell death, including apoptosis, a programmed cell-death process.
At the same time, the affected neurons produced lower levels of molecules linked to normal nerve-cell work, Stanford said. Those included molecules involved in growing and guiding axons, forming contacts with other cells and sending chemical signals to muscle fibers and other targets.
Protective signals appeared too late
The researchers also examined transcription factors, proteins that control which molecules a cell makes and in what amounts, according to Stanford. The team expected the changes in these regulators to mostly drive degeneration, Gautier said through the university.
Instead, the study found increases in transcription factors that appeared to be protective, Stanford said. Gautier said those responses may represent a defense mounted by the neuron, but one that arrives too late or is too weak to prevent cell death.
The team also tested spinal cord tissue from ALS patients, according to Stanford. Those experiments suggested that similar molecular changes occur in humans, supporting the relevance of the mouse findings to human disease.
Stanford said the results remain an early step toward treatment. Gautier and colleagues plan to delete or suppress genes linked to cell death and dysfunction to test whether that slows disease progression in mice.
Another possible route would be to develop therapies that enhance the protective responses seen in the vulnerable motor neurons, Stanford said. Gautier said the aim is to help those cells withstand ALS-related damage for longer.
This story draws on original reporting from Medical Xpress.