
Researchers add experimental data to AlphaFold to model moving proteins
An ISTA-led team says experiment-guided AlphaFold can predict ensembles of protein shapes instead of one dominant structure.
By Tom Brennan · Health & Medicine Correspondent
3 min read
Researchers have modified AlphaFold so the AI system can use experimental measurements to model multiple protein shapes, not just a single likely structure. The work matters because many proteins function through motion, while major structure databases and prediction tools often present them as fixed objects.
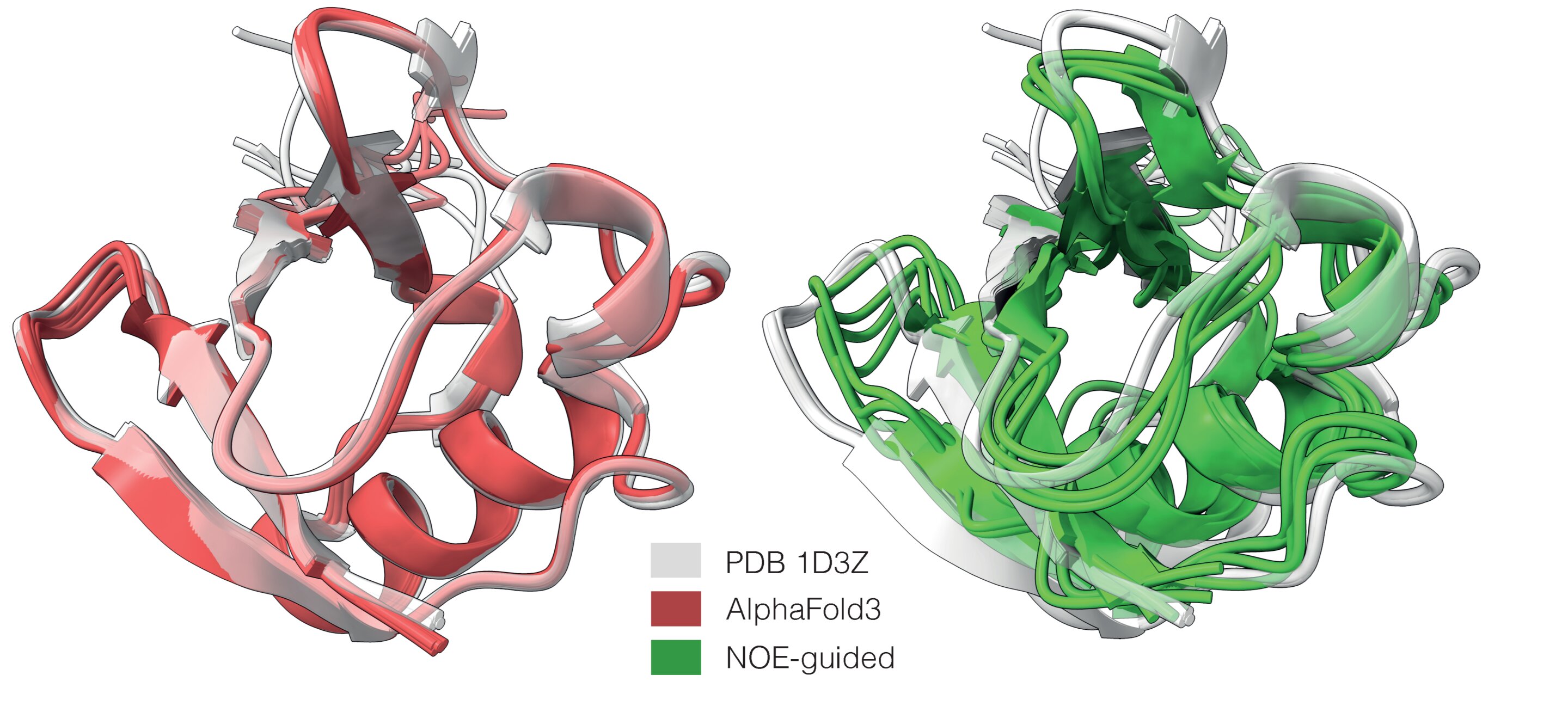
The Institute of Science and Technology Austria said the approach, developed with international collaborators and published in Nature Biotechnology, guides AlphaFold3 with data such as nuclear magnetic resonance measurements and cryo-electron microscopy maps. According to ISTA, the method produces protein ensembles that better match experimental evidence in cases where standard AlphaFold3 collapses a flexible molecule into one dominant conformation.
A push beyond static protein pictures
AlphaFold has become a central tool in structural biology by predicting three-dimensional protein structures from amino acid sequences. ISTA said the system, like other models, was trained heavily on static structures from X-ray crystallography, which make up about 85% of the Protein Data Bank.
Those crystal structures have been essential to biology, medicine and drug research, according to ISTA, but they can understate how much proteins move. Flexible regions, including loops that may be missing from crystallographic models, have often been treated as indistinct connectors even though the team says they can carry biological meaning.
Alex Bronstein, an ISTA professor who co-led the work, said proteins are dynamic molecules and that modeling their motion could show why those movements matter for function. The project was led by Bronstein with Ailie Marx of Tel-Hai University and MIGAL—Galilee Research Institute, ISTA professor Paul Schanda, and Sanketh Vedula of Princeton University and the Broad Institute, according to ISTA.
Experimental data as a guide
AlphaFold already uses evolutionary information, including relationships among residues that appear to have changed together, ISTA said. The researchers argue that current systems apply that information too rigidly and do not fully reflect the sequence and structural context of each protein.
The experiment-guided version is designed to extract subtler information about structure and dynamics from sequences and measurements. Advaith Maddipatla, an ISTA doctoral student and first author of the study, said the team is building a model that can capture details that leading protein databases, including the PDB, cannot represent in their current form.
ISTA said the model was tested on examples including ubiquitin, leech decorsin and an amyloid-beta fibril. In those cases, the researchers reported that experimental guidance helped generate structures consistent with measurements, including when AlphaFold3 missed key structural features.
From proof of concept to research tool
The team’s longer-term goal is to use each newly modeled structure to improve later predictions, ISTA said. Bronstein said the approach could help organize structural information in the PDB within a few years by making models more consistent with physical measurements.
The researchers also want the system to reconstruct plausible protein ensembles from noisy data. Bronstein said the blur that static crystallography often treats as a problem is the signal the group wants to recover.
According to ISTA, the method could eventually help model large protein complexes and support inverse protein design, a drug discovery and bioengineering approach that designs sequences to fold into chosen structures. Schanda said some proteins pass quickly through conformations that are important to their function, and the team wants the model to capture those states.
ISTA said an earlier version of the study was presented at the 2025 International Conference on Machine Learning. A follow-up study on speeding the model’s inference time has been accepted to ICML 2026, and a separate bioRxiv preprint applies the method to β2-microglobulin conformations that the researchers say conventional refinement missed.
This story draws on original reporting from Phys.org.