
AI model predicts protein binding sites atom by atom
Researchers say Void-X can generate atomic details of protein interfaces, a step that could aid drug discovery and synthetic biology.
By Tom Brennan · Health & Medicine Correspondent
3 min read
Researchers at the Shanghai Institute of Organic Chemistry have developed a generative AI model that predicts how proteins fit together at atomic scale. The work matters because protein interactions underpin many drug targets, including therapies built around antibodies, insulin and other protein-based treatments, according to the Chinese Academy of Sciences.
The model, called Void-X, was described in a study published June 9 in Proceedings of the National Academy of Sciences. The Chinese Academy of Sciences said the system designs protein interfaces by filling in missing atomic structures within contact regions, rather than starting with a whole protein framework.
A bottom-up approach to protein design
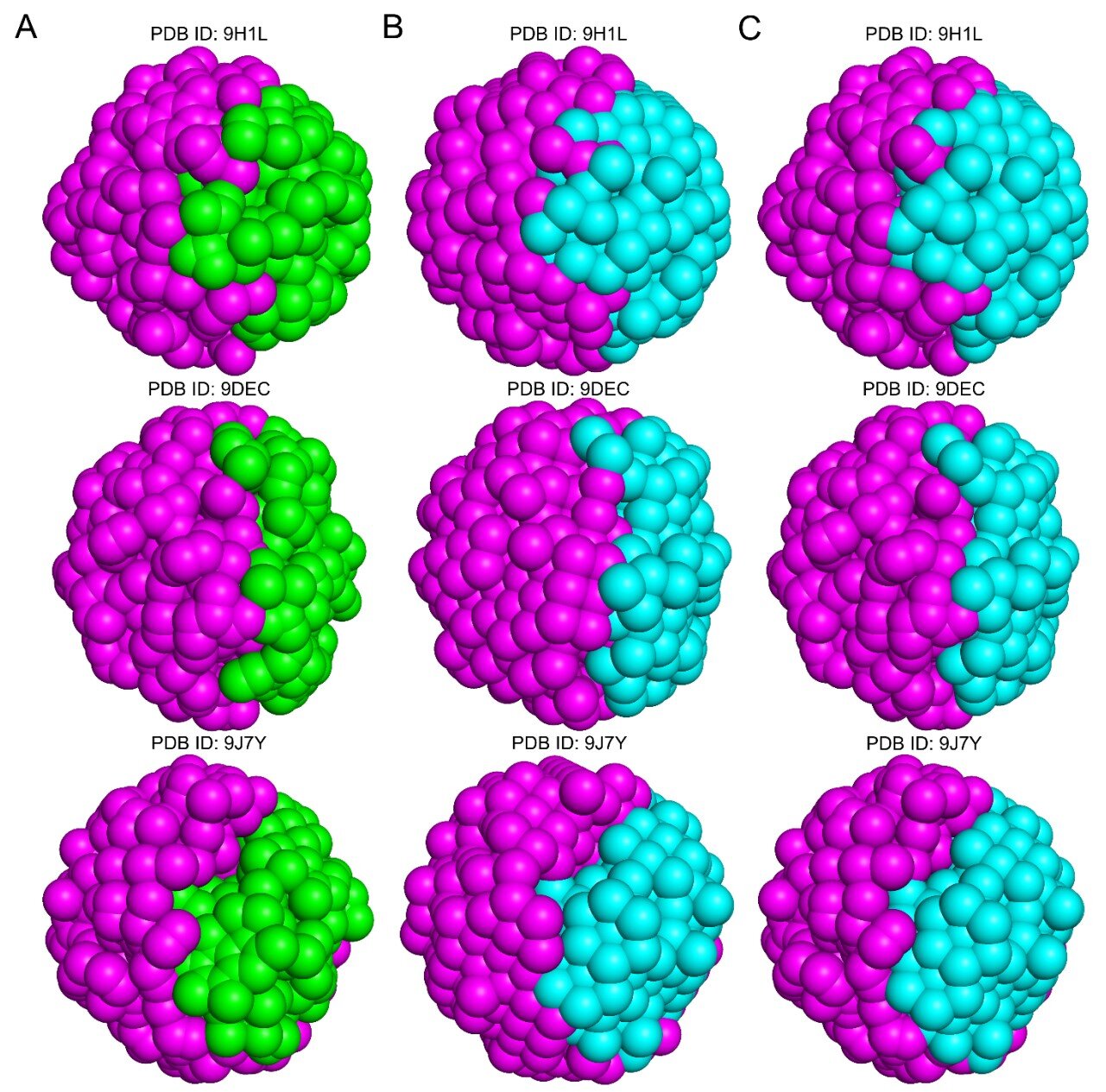
Many AI protein-design methods first create a broad scaffold that can sit on a target site, then search for amino acid sequences that improve binding, according to the researchers. Void-X instead works from local atomic packing: it learns patterns among nearby atoms and uses those patterns to infer atoms that should occupy gaps in a protein interface.
The study describes this as an atomic filling model. In that setup, known atoms around a region act as the prompt, and the model predicts masked atoms inside nearby space. The researchers say this lets the system generate tightly packed atomic clusters for defined structural regions.
That approach is meant to reflect how stable macromolecular complexes form, according to the study. The model accounts for close-range interactions among neighboring atoms as well as couplings involving atoms farther away.
Training on protein structures
Yang Jing, Yuan Junying and James J. Chou built the training set from experimentally resolved structures in the Protein Data Bank, according to the Chinese Academy of Sciences. The data set included more than 8 million spherical clusters of atoms.
For each cluster, the researchers masked about 30% of the atoms on the outer edge that were also spatially connected, the academy said. The unmasked atoms were left as context for the model to use while predicting the missing portion.
Void-X has 172 million parameters, according to the study. The researchers reported prediction accuracy of 78.3% for atomic clusters within a single protein chain and 68.2% for clusters between separate chains.
Possible uses in medicine and biology
The Chinese Academy of Sciences said the work could help expand methods for designing biomolecular interfaces. Protein-protein interactions are central to many biological processes, including tissue formation, molecular transport, cell communication and immune defense, according to the academy.
The researchers say better prediction and engineering of these interactions could support therapeutic development. They also said such work could complement advances in delivery systems, including adeno-associated virus platforms and mRNA lipid nanoparticles.
According to the study, Void-X offers a route for de novo generation of protein interactions at atomic resolution. The researchers said the model could have applications in drug discovery, synthetic biology and related fields.
This story draws on original reporting from Phys.org.